
Apartados
Resumen
Abstract
Introducción
Discusión
Referencias
Síndrome de Angelman: Serie de Casos y Revisión de la Literatura
Angelman Syndrome: Case Series and Review
1José Óscar Sánchez-Rendón, 2Gabriel Mancera-Elías, 3Josué Israel Cervantes, 4Miguel Ángel Cerón-Serralta.
1Médico Anestesiólogo Pediatra, Jefe de Anestesiología del Hospital del Niño Morelense y práctica privada. 2Médico Anestesiólogo Pediatra, adscrito al Instituto Nacional de Pediatría. 3Médico Anestesiólogo Pediatra, adscrito al Hospital del Niño Morelense. 4Médico Residente de tercer año del Hospital Regional ISSSTE Monterrey, Nuevo León, México.
Anestesia en México 2025; 37(3): 288–295 https://doi.org/10.64221/aem-37-3-2025-034
Fecha de recepción junio 2025, fecha de revisión julio 2025, fecha de publicación septiembre 2025.
oscarsanren@gmail.com
Resumen
El Síndrome de Angelman (SA)es un trastorno genético que presenta desafíos en el manejo anestésico debido a su compleja presentación clínica y genética. Este estudio busca revisar retrospectivamente los casos de pacientes pediátricos con SA sometidos a procedimientos anestésicos, y complementar con una revisión de la literatura para ofrecer una visión completa de los desafíos y estrategias en el manejo anestésico de estos pacientes. Material y métodos: Se revisaron retrospectivamente los historiales de pacientes con SA tratados anestésicamente entre 2012 y 2022. Se analizaron datos demográficos, manejo anestésico y complicaciones. Además, se realizó una revisión sistemática de la literatura utilizando bases de datos como PubMed, Google Scholar y EBSCO. Resultados: Se identificaron cinco pacientes que recibieron anestesia durante este periodo, abarcando un total de siete procedimientos. Los desafíos más comunes incluyeron manejo de la vía aérea, eventos de bradicardia y control del dolor postoperatorio. No se encontraron complicaciones mayores en el manejo anestésico. Conclusiones: El Síndrome de Angelman (SA) exige manejo anestésico especial por una potencial vía aérea difícil (macroglosia/prognatismo), epilepsia (ajustar fármacos por interacciones), hipertonía vagal (riesgo de bradicardia). La analgesia multimodal es clave para optimizar el confort postoperatorio. Palabras clave: Síndrome de Angelman, manejo anestésico y complicaciones anestésicas.
Abstract
Introduction: Angelman Syndrome (SA) is a genetic disorder that presents challenges in anesthetic management due to its complex clinical and genetic presentation. This study aims to retrospectively review pediatric patients with Angelman Syndrome undergoing anesthetic procedures, and complement with a literature review to provide a comprehensive view of the challenges and strategies in the anesthetic management of these patients. Materials and Methods: A retrospective review of medical record of patients with Angelman Syndrome treated with anesthesia from 2012 to 2022 was conducted. Demographic data, anesthetic management, and complications were analyzed. Additionally, a systematic literature review was performed using databases such as PubMed, Google Scholar, and EBSCO. Results: Five patients were identified who received anesthesia during this period, covering a total of seven procedures. Common challenges included airway management, bradycardic events, and postoperative pain control. No major complications were found in anesthetic management. Conclusions: Angelman Syndrome (AS) requires special anesthetic management due to a potentially difficult airway (macroglossia/ prognathism), epilepsy (adjust drugs for interactions), vagal hypertonia (risk of bradycardia). Multimodal analgesia is key to optimizing postoperative comfort. Keywords: Angelman Syndrome, anesthetic management and anesthetic complications.
Introducción:
El Síndrome de Angelman (SA) es una entidad genética cuya manifestación clínica ha despertado la atención de la comunidad médica a lo largo de las décadas. No presenta una prevalencia específica entre hombres y mujeres. La incidencia reportada oscila entre uno en 10,000 y uno en 52,000 (1,2). A pesar de esta estimación, es posible que el síndrome puede estar subdiagnosticado debido a la variabilidad en su presentación clínica, encontrando casos cuya detección se prolonga hasta la adolescencia (1). Se presenta como un trastorno genético caracterizado por retraso en el neurodesarrollo, anormalidades craneofaciales, ataxia cerebelosa, convulsiones, alteraciones en el sueño y comportamiento idiosincrático (risa incontrolada, apariencia feliz, alteraciones del habla, movimientos repetidos) (2). El síndrome fue descrito por primera vez en 1965 por el pediatra Harry Angelman. Se le denominó originalmente “Síndrome del Títere Feliz». Sin embargo, con el tiempo, esta nomenclatura se transformó, tomando el apellido del pediatra, para convertirse en SA (3). Respecto a su patogénesis y factores de riesgo, se ha encontrado una asociación entre el SA y la edad materna avanzada, así como con bebés concebidos mediante técnicas de reproducción asistida (1). Los pacientes diagnosticados con SA a menudo se enfrentan a desafíos anestésicos únicos, debido a su falta de cooperación como la irritabilidad y un comportamiento errático. Debido a la complejidad y los desafíos específicos asociados con el SA en el contexto anestésico, resulta crucial que los anestesiólogos posean un conocimiento profundo y actualizado de este síndrome antes de abordar cualquier caso. Para contribuir a este campo de conocimiento hemos realizado una revisión retrospectiva de todos los pacientes con SA que fueron manejados anestésicamente en nuestra práctica durante los últimos 10 años. Además, para complementar nuestra revisión institucional, hemos llevado a cabo una revisión sistemática de la literatura. Cuya intención es recopilar y sintetizar las actualizaciones de este tema y también añadir nuestra experiencia de manejo en pacientes con síndrome de Angelman.
Material y métodos
Este estudio retrospectivo se incluyeron pacientes cuyos representantes legales proporcionaron autorización para el uso de sus historiales médicos. Los pacientes fueron identificados con SA y tratados en nuestra institución y práctica privada, habiendo tenido algún procedimiento anestésico desde el primero de enero de 2012 hasta el 31 de diciembre de 2022. Realizamos una revisión de los expedientes electrónicos, abarcando datos demográficos, comorbilidades, manejo anestésico (reporte de bradiarritmias, manejo de vía aérea, convulsiones posoperatorias y estrategias de analgesia), procedimientos y complicaciones quirúrgicos intraoperatorias. La revisión de literatura se llevó a cabo en las bases de datos de PubMed (2000-presente), Google Scholar (2000-presente) y EBSCO (2000-presente), utilizando las siguientes palabras clave: Angelman, Síndrome de Angelman, anestesia, complicaciones posoperatorias y cirugía.
Resultados
Cinco pacientes fueron identificados que recibieron anestesia durante este periodo. Estos pacientes presentaron siete procedimientos; siete realizados con anestesia general. Dos pacientes reportaron resultados de fish 15q11,2q13 positivo, el resto fue diagnosticado por criterios clínicos. Las variables demográficas fueron presentadas en la (Tabla I). Todos los pacientes tuvieron retraso moderado a grave en el neurodesarrollo. Los procedimientos y variables anestésicas son resumidas en la (Tabla II). El curso clínico de todos pacientes fue sin complicaciones, solo con algunas observaciones:
Discusión
El SA es una condición genética rara pero bien definida, caracterizada por una compleja etiología genética. Este síndrome se asocia principalmente con anomalías en el cromosoma 15q11.2-q13, una región crítica que afecta la expresión del gen UBE3A. Las etiologías conocidas del SA son responsables del silenciamiento de este gen e incluyen (5):
Las mutaciones en el receptor GABA-A tienen un impacto significativo en la respuesta a ciertos medicamentos, especialmente los anestésicos. Los pacientes con estas mutaciones pueden presentar respuestas impredecibles a anestésicos intravenosos que interactúan con GABA-A. Un ejemplo concreto de esto es una sensibilidad elevada a las benzodiacepinas. Además, ciertos medicamentos que actúan en el sistema GABA podrían no ser efectivos en estos individuos. Un caso ilustrativo es el midazolam, que en algunos pacientes no ha demostrado ser eficaz debido a estas variabilidades genéticas (7). En modelos animales, se ha relacionado a los receptores N-metil- D-aspartato (NMDA) con trastornos de UBE3A, donde UBE3A representa la ligasa de ubiquitina-proteína E3A. A diferencia del UBE3A paterno, el materno está activo en el cerebelo y el hipocampo (4). A principios de 2010, se identificó un sustrato de UBE3A llamado “Arc”. Arc es una proteína sináptica que regula la internalización de otro receptor de glutamato, el receptor de ácido α-amino-3-hidroxi-5-metil-4-isoxazolpropiónico (AMPA). Como resultado de estas investigaciones en modelos animales, se hipotetiza que las disfunciones cognitivas en pacientes con AS podrían deberse a disrupciones de los receptores NMDA o AMPA [4]. Estas relaciones entre las mutaciones genéticas, la función del receptor GABA-A y la respuesta a medicamentos resaltan la intrincada red de interacciones que subyace en el sistema nervioso y en el comportamiento humano. Es vital considerar estas interacciones al abordar trastornos como el Síndrome de Angelman, para garantizar un tratamiento adecuado y personalizado para cada paciente (7). Un estudio de Li et al. ha arrojado luz sobre la anestesia en su capacidad para bloquear selectivamente la consciencia sin afectar otras actividades cerebrales no conscientes, esto obtenido al examinar el isótopo de xenón con propiedad cuántica de espín nuclear ½. El xenón con espín nuclear ½ es menos potente como anestésico en comparación con otros isótopos de xenón sin espín, a pesar de tener acciones químicas idénticas. Esto ha llevado a la sugerencia de que el espín nuclear del xenón puede antagonizar su propia acción anestésica al promover la consciencia (8). Este hallazgo respalda una categoría de teorías conocidas como “consciencia cuántica”. Las teorías de consciencia cuántica implican fenómenos físicos que ocurren en escalas muy pequeñas, pero que pueden tener implicaciones a gran escala. Estos incluyen efectos de campo, entrelazamiento no local (donde partículas separadas están conectadas en espacio y tiempo), coherencia (la condensación de múltiples partículas en entidades unitarias) y superposición cuántica (que se utiliza en la computación cuántica) (9). El síndrome de Angelman, caracterizado por una dinámica neural que se asemeja al sueño durante la vigilia, presenta un desafío intrigante para la comprensión de la consciencia. A través de estudios de marcadores de encefalograma (EEG), se ha descubierto que los patrones de actividad cerebral en individuos con SA son únicos y se disocian de lo que normalmente asociamos con la vigilia consciente (10). Una evidencia ampliamente aceptada proviene del sueño de ondas lentas, la anestesia, el coma y las crisis epilépticas que relacionan la actividad lenta y de alto voltaje del EEG con la pérdida de conciencia. En niños con el síndrome de Angelman (SA) a pesar de estar completamente despiertos y mostrar comportamiento volitivo, muestran un fenotipo de EEG de frecuencia delta hipersincrónica (1-4 Hz) que es típico de la inconsciencia (11). En este contexto, el SA se ha convertido en un punto focal para cuestionar y estudiar las tradicionales métricas y entendimientos de la conciencia. La oscilación delta, específicamente su valle, se asocia con estados en los que las neuronas corticales están silenciadas. La presencia de comportamiento volitivo y vigilia (dificultad para dormirse) en SA, a pesar de estos ritmos delta difusos, introduce un paradigma intrigante. Esto sugiere que, incluso en condiciones de aparente inconsciencia según las medidas tradicionales de EEG, existen complejas dinámicas cerebrales que desafían nuestra comprensión convencional de la conciencia (12). Es vital profundizar en la comprensión de la enfermedad para optimizar la atención al paciente. En este sentido, estudios recientes han mostrado que las características específicas del electroencefalograma (EEG) en pacientes con SA pueden ofrecer valiosa información sobre la consciencia y su relación con una dinámica cortical anormal (12). Estos hallazgos no sólo enriquecen la comprensión del SA, sino que también abren puertas a investigaciones futuras en el ámbito de la neurología y la consciencia.
Consideraciones Anestésicas: Discapacidad intelectual:
intelectual profunda y suelen tener dificultades de comunicación. Aunque son generalmente de buen carácter, su discapacidad cognitiva y habilidades comunicativas dificultan su cooperación durante los procedimientos médicos. En consecuencia, a menudo requieren sedación, incluso para procedimientos menores. La disponibilidad de fármacos para sedación, como la dexmedetomidina y el midazolam a dosis convencionales, son una buena opción en el manejo de los pacientes pediátricos, aunque se debe considerar que en los niños con síndrome de Angelman no siempre se obtendrá el resultado esperado debido a sus defectos en el sistema GABA, lo que puede afectar la eficacia de la sedación. El principal reto es en la evaluación del dolor postoperatorio debido a su dificultad de comunicación. Como alternativa a la Escala Numérica de Dolor (ENA), las escalas CRIES o FLACC pueden ser utilizadas para evaluar el dolor en niños no cooperadores. También se describieron perturbaciones del sueño, aunque no se describen sus efectos postoperatorios o asociación relacionada a emersión anestésica retardada (15).
Anomalías en el receptor GABA-A:
Además de las anomalías en el gen UBE3A, en algunos pacientes el defecto genético puede afectar el gen que codifica la subunidad 3 del receptor GABA-A. Esto podría influir en las características neurológicas del síndrome de Angelman, como las convulsiones y las anormalidades de comportamiento. Puede haber una respuesta impredecible a los anestésicos intravenosos que actúan a través de interacciones con GABA-A. La modulación por drogas como los benzodiazepínicos, que se utilizan por sus efectos sedantes/hipnóticos y ansiolíticos, depende de la composición de subunidades de los receptores GABA-A, lo que subraya la importancia de la diversidad de receptores en la farmacología anestésica. En nuestra experiencia, algunos pacientes han requerido dosis adicionales de hipnóticos para entrar a plano anestésico, también hemos observado que los tiempos de inducción inhalatoria con sevoflurano son más prolongados, lo anterior posiblemente asociado con las anomalías genéticas descritas (16).
Actividad Convulsiva
Los pacientes diagnosticados con el síndrome de Angelman suelen exhibir patrones electroencefalográficos característicos, con aproximadamente el 80 % experimentando actividad convulsiva. En esta serie, tres pacientes tenían trastornos convulsivos diagnosticados, mientras que dos mostraron electroencefalograma normal. No se encontró evidencia que sugiriera un empeoramiento de la actividad convulsiva después de la anestesia. Es crucial que los anestesiólogos aseguren que los regímenes antiepilépticos no se interrumpan durante el período perioperatorio (17).
Anomalías Craneofaciales
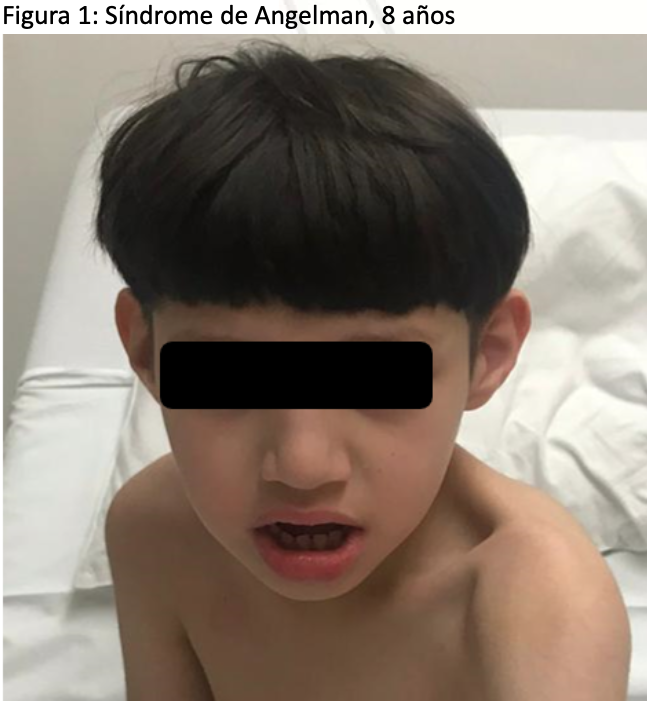
En un estudio comprensivo de 226 casos de Brukiewicz se buscó identificar las particularidades craneofaciales y orales de los pacientes con SA. Los hallazgos demuestran una alta prevalencia de microcefalia postnatal, evidente alrededor de los dos años hasta en un 52.21 % de los casos, microbraquicefalia, el prognatismo mandibular y la macrostomía en 33.63 %, junto con la macroglosia y anomalías como el paladar ojival alto e hipoplasia de los huesos maxilares, aunque menos comunes, presentes en menos del 1 % de los casos, podrían complicar tanto la ventilación con máscara facial como el acceso para la intubación endotraqueal. Estos datos, extraídos de una revisión literaria exhaustiva de 16 publicaciones seleccionadas de bases de datos confiables, enfatizan la necesidad de una evaluación preoperatoria meticulosa y la preparación para técnicas de intubación adaptadas o avanzadas (Figura 1) (18).
Aumento del Tono Vagal
La hipertonía vagal en estos pacientes puede conducir a complicaciones significativas durante la anestesia, incluyendo episodios bradicárdicos profundos y reacciones vágales extremas, algunas de las cuales han sido desencadenadas por episodios de risa. Este fenómeno destaca la importancia de un cuidado anestésico meticuloso y adaptado a las necesidades específicas de los pacientes con SA (19). Además, se han reportado casos en los que la hipertonía vagal ha desempeñado un papel crítico en eventos adversos graves. Por ejemplo, se ha documentado un caso de asistolia durante un procedimiento de laparoscopia en un niño con síndrome de Angelman, lo que subraya el riesgo potencial asociado con el predominio del tono vagal en estos pacientes. Este riesgo es especialmente relevante en el contexto de la implantación de un estimulador del nervio vago en pacientes con síndrome de Angelman. Las crisis de risa, comunes en individuos con este síndrome, pueden precipitar eventos como síncope, bradicardia profunda y asistolia, secundarios a la hipertonía vagal. Por lo tanto, sugerimos la medicación con un agente anticolinérgico, como atropina, antes de la inducción y durante las maniobras que puedan estimular el centro vagal (20).
Anomalías Musculares
La hipotonía truncal es una característica observada comúnmente durante la infancia en pacientes con síndrome de Angelman, que con el tiempo puede evolucionar hacia un aumento del tono muscular en las extremidades. Este cambio en la presentación de los síntomas plantea preocupaciones acerca de la seguridad en el uso de relajantes musculares durante procedimientos que requieren anestesia. No obstante, las evidencias de una serie de casos han mostrado que el uso de estos medicamentos, en su mayoría, no ha derivado en incidentes adversos. Además, se ha identificado la distonía como un hallazgo clínico común y previamente no reconocido entre adolescentes y adultos afectados por este síndrome [2] (21).
Complicaciones respiratorias
Aunque estas no son características principales del síndrome de Angelman, se han reportado casos que presentan graves problemas respiratorios postoperatorios. La relación directa entre estas complicaciones y el síndrome aún no está clara, planteando la pregunta de si son inherentes al síndrome de Angelman o si representan riesgos generales asociados con la anestesia en este grupo de pacientes. Un artículo notable describe a un paciente de cinco años con un diagnóstico establecido de SA, quien falleció debido a una obstrucción de las vías respiratorias superiores ocasionada por amígdalas masivamente agrandadas, complicación de una mononucleosis infecciosa. Este caso subraya los desafíos para evaluar la gravedad de las enfermedades subyacentes en niños con retraso en el desarrollo, especialmente cuando no pueden vocalizar síntomas de empeoramiento y angustia. Además, los signos de estrechamiento de las vías respiratorias superiores debido a infecciones pueden ser menos aparentes en pacientes con síndrome de Angelman, en parte debido a las dificultades para succionar y tragar, lo que resulta en comportamientos de babeo y masticación excesiva (22). En otro caso, se detalla la cirugía de un paciente con síndrome de Angelman severo que necesitaba una operación de separación laringotraqueal, un procedimiento poco común en pacientes con este diagnóstico. Este escenario destaca la importancia de utilizar pruebas de hibridación genómica comparativa para confirmar el tamaño de la deleción y, por ende, la severidad clínica del síndrome en el paciente. Tal evaluación genética puede proporcionar información valiosa para anticipar y manejar complicaciones específicas relacionadas con el síndrome (23).
Conclusión
El Síndrome de Angelman (SA) requiere un enfoque anestésico especializado debido a sus características únicas. Los pacientes presentan rasgos faciales distintivos como macroglosia y prognatismo que pueden complicar el manejo de la vía aérea, La epilepsia, presente en la mayoría de casos, exige conocer posibles interacciones farmacológicas con medicamentos anticonvulsivantes. La hipertonía vagal característica del SA puede manifestarse como bradicardia intraoperatoria. Es importante destacar que los pacientes con SA pueden presentar una disociación entre su estado clínico y los patrones EEG, mostrando actividad delta típica de sueño profundo mientras permanecen conscientes. (Entropía, BIS, etc).
")
")
Referencias
- Bevinetto CM, Kaye AD. Perioperative considerations in the patient with Angelman syndrome. J Clin Anesth. 2014;26(1):75-9. DOI: 10.1016/j.jclinane.2013.07.015
- Warner ME, Martin DP, Warner MA, Gavrilova RH, Sprung J, Weingarten TN. Anesthetic Considerations for Angelman Syndrome: Case Series and Review of the Literature. Anesth Pain Med. 2017;7(5):e57826. DOI: 10.5812/aapm.57826
- Campero L. Happy Puppet Syndrome: A Case Report of Anesthetic Management. AANA J. 2018;86(1):67-71. PMID: 31573496
- Witte W, Nobel C, Hilpert J. Anästhesie beim Angelman-Syndrom. Anaesthesist. 2011;60(7):633-40. DOI: 10.1007/s00101-011-1873-4
- Cascella M, et al. Pathophysiology of Nociception and Rare Genetic Disorders with Increased Pain Threshold or Pain Insensitivity. Pathophysiology. 2022;29(3):435-452. DOI: 10.3390/pathophysiology29030035
- Orphanet. Síndrome de Angelman. Disponible en: orpha.net. [Acceso: 2 de octubre de 2023]
- Gupta N, et al. Genetic heterogenicity of Angelman syndrome and its significance to the anesthesiologist. Saudi J Anaesth. 2015;9(1):105-106. DOI: 10.4103/1658-354X.146336
- Li N, et al. Nuclear Spin Attenuates the Anesthetic Potency of Xenon Isotopes in Mice. Anesthesiology. 2018;129(2):271-277. DOI: 10.1097/ALN.0000000000002226
- Hameroff SR. Anesthetic Action and “Quantum Consciousness”: A Match Made in Olive Oil. Anesthesiology. 2018;129(2):228-231. DOI: 10.1097/ALN.0000000000002273
- Frohlich J, et al. Neural complexity is a common denominator of human consciousness. Commun Biol. 2022;5(1):1374. DOI: 10.1038/s42003-022-04331-7
- Frohlich J, et al. High-voltage, diffuse delta rhythms coincide with wakeful consciousness in Angelman syndrome. Neurosci Conscious. 2020;2020(1):niaa005. DOI: 10.1093/nc/niaa005
- Huang Z. Temporospatial Nestedness in Consciousness. Entropy. 2023;25(7):1074. DOI: 10.3390/e25071074
- Williams CA, et al. Angelman syndrome 2005: updated consensus for diagnostic criteria. Am J Med Genet A. 2006;140(5):413-8. DOI: 10.1002/ajmg.a.31074
- Wheeler AC, et al. Unmet clinical needs and burden in Angelman syndrome. Orphanet J Rare Dis. 2017;12(1):164. DOI: 10.1186/s13023-017-0716-z
- Thibert RL, et al. Neurologic manifestations of Angelman syndrome. Pediatr Neurol. 2013;48(4):271-9. DOI: 10.1016/j.pediatrneurol.2012.09.015
- Sigel E, Steinmann ME. Structure, function, and modulation of GABA(A) receptors. J Biol Chem. 2012;287(48):40224-31. DOI: 10.1074/jbc.R112.386664
- Santini E, Klann E. Genetically Dissecting Cortical Neurons Involved in Epilepsy in Angelman Syndrome. Neuron. 2016;90(1):1-3. DOI: 10.1016/j.neuron.2016.03.023
- Brukiewicz K, Komisarek O. Craniofacial disorders in the course of Angelman syndrome. J Educ Health Sport. 2020. DOI: 10.12775/jehs.2020.10.06.015
- Gardner J, et al. Vagal hypertonia and anesthesia in Angelman syndrome. Paediatr Anaesth. 2008. DOI: 10.1111/j.1460-9592.2008.02487.x
- Timko NJ, Marshall JM. Anesthetic considerations for vagal nerve stimulator implantation in Angelman syndrome. Paediatr Anaesth. 2023;33(10):874-875. DOI: 10.1111/pan.14702
- Ferlazzo E, et al. Dystonia in Angelman syndrome: a common, unrecognized clinical finding. J Neurol. 2021;268:2208-2212. DOI: 10.1007/s00415-020-10395-4
- Herbst J, Byard R. Sudden Death and Angelman Syndrome.